Angelo Cignarelli, Valentina Annamaria Genchi, Sebastio Perrini, Annalisa Natalicchio, Luigi Laviola, Francesco Giorgino
Dipartimento dell’Emergenza e dei Trapianti di Organo, Sezione di Medicina Interna, Endocrinologia, Andrologia e Malattie Metaboliche, Università degli Studi di Bari “Aldo Moro”
INTRODUZIONE
La ricerca biomedica degli ultimi dieci anni ha consentito di incrementare le nostre conoscenze sul ruolo del tessuto adiposo (TA) in condizioni fisiologiche e patologiche. Il TA è riconosciuto come un organo a tutti gli effetti con funzioni metaboliche ed endocrine altamente attive, giocando un ruolo centrale nella regolazione dell’omeostasi energetica attraverso le sue funzioni a livello locale e sistemico. Da un lato, il TA accumula e libera energia sotto forma di lipidi in base alle differenti richieste metaboliche (Fig. 1),

dall’altro agisce come organo endocrino producendo numerosi fattori bioattivi, noti come adipochine, in grado di comunicare con altri organi e modulare un’ampia gamma di segnali che regolano importanti funzioni, tra cui quella immunitaria, endocrina, rigenerativa e meccanica (Tab. 1).
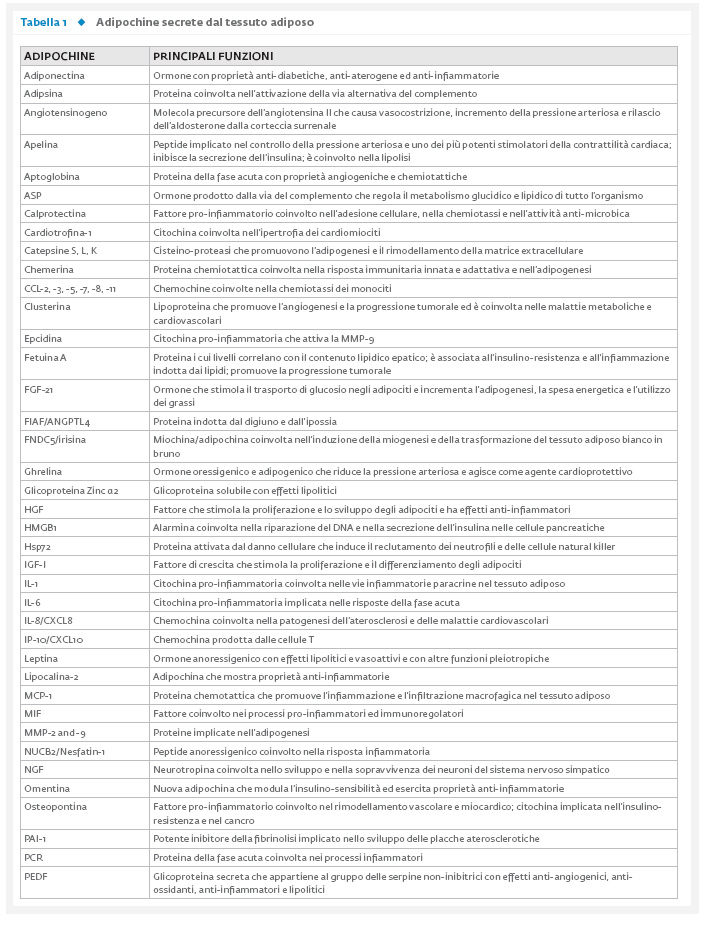
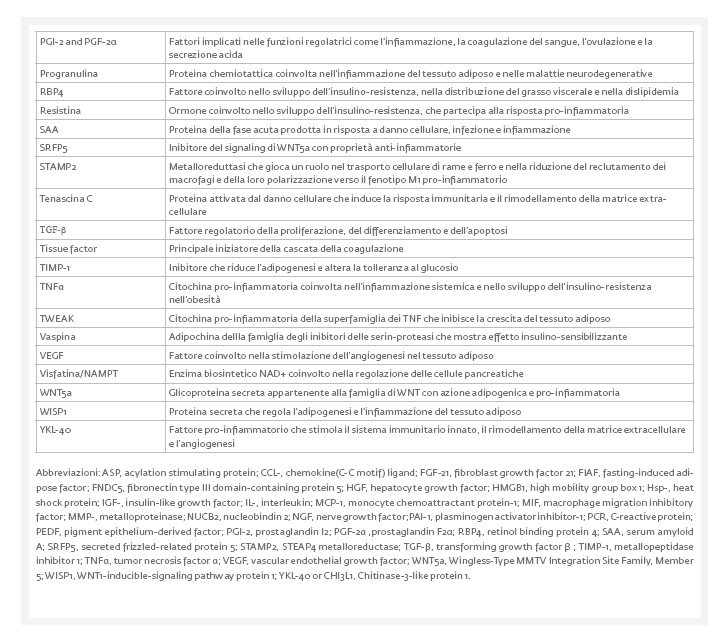
Inoltre, il TA, in determinate condizioni e sotto opportuni stimoli, è anche in grado di ossidare i lipidi e/o di dissipare energia sotto forma di calore al fine di mantenere l’eutermia.
Gli studi che hanno indagato i meccanismi di regolazione dell’espansione del TA hanno chiarito, sebbene non del tutto, le modalità attraverso cui si realizza l’incremento ponderale e, soprattutto, lo sviluppo dei suoi disordini metabolici correlati come insulino-resistenza, patologie cardiovascolari e diabete mellito di tipo 2; numerose evidenze, infatti, indicano come la disfunzione del TA sia uno dei primi meccanismi alla base dello sviluppo e/o progressione dell’insulino-resistenza e del diabete. Soggetti affetti da obesità spesso presentano adipociti aumentati di volume con una ridotta capacità di ulteriore espansione e di accumulo di energia sotto forma di lipidi, esponendo altri tessuti ad un eccessivo flusso di lipidi. Inoltre, l’eccessiva ipertrofia adipocitaria si accompagna ad una riduzione del flusso ematico per un insufficiente apporto vascolare, con conseguente ipossia cellulare, alterazione della secrezione delle adipochine, apoptosi, infiltrazione macrofagica ed infiammazione a livello del TA. Tutti questi elementi concorrono nel determinare una conversione del TA da organo sano ad organo infiammato in grado di indurre insulino-resistenza e, conseguentemente, diabete mellito di tipo 2.
In questa rassegna saranno presi in considerazione alcuni elementi di fisiologia e fisiopatologia del TA con particolare riferimento all’associazione tra TA disfunzionale, insulino-resistenza e sviluppo dell’iperglicemia.